New Regulation of medical devices
Author: TIC Salut Social / 9 of March of 2020

9 of March of 2020
The new Regulation of medical devices is ready to enter into force. The next 26th of May 2020 the Regulation (EU) 2017/745 will be focused on the following medical devices:
- Class III emplementable products.
- Products that use human tissues.
- Combined products (those that include a part of medicine and another of healthcare product) according to the 117 Article.
- Products of the Annex XVI without medical purposes, and now lasers, squads of liposuction, etc.
Why these changes?
The Regulation will replace at the current 93/42/EEC (MDD) (of medical devices) and the 90/385/EEC (AIMDD).
These new Regulations are a important step beyond and they are entering into force across the European Union.
Image 1. Regulations and Directors
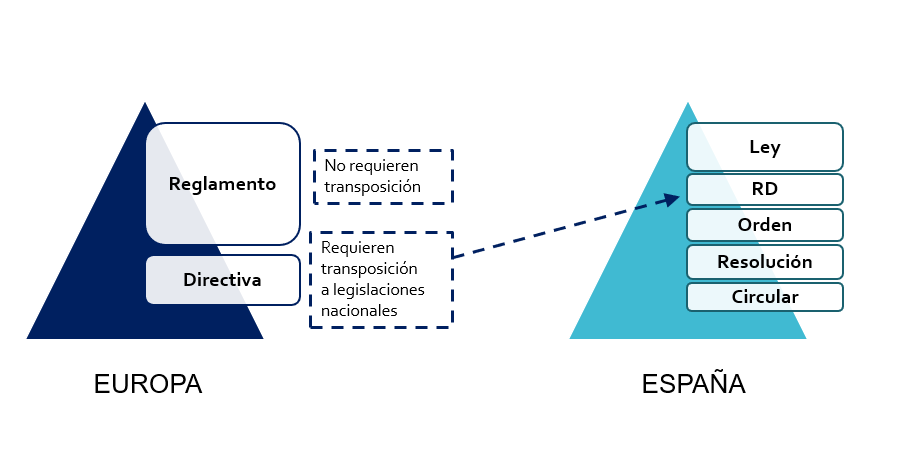
Source: CSV Experts
Image 2: Valid legislation at Europa
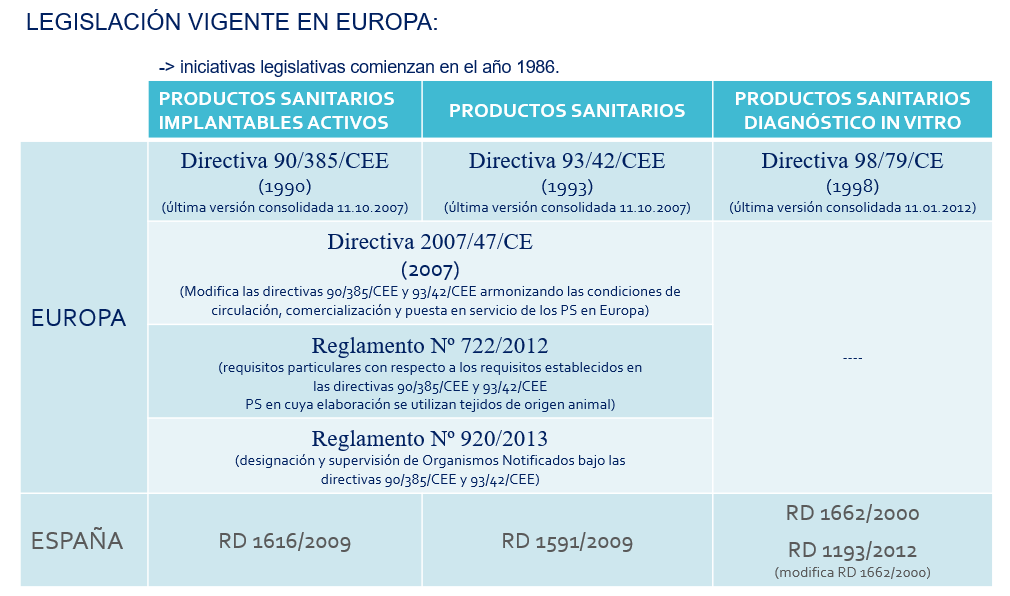
Source: CSV Experts
According to the MedTech Europe, the medical tech features for a constant innovation, as a result of I+D investment, healthcare products tend to need among 17-24 months to be reñeased in the market. The following figures can give us an idea: in 2017, the patents’ office registered more than 13.000 requests of patents for medical tech (7,9% of the total amount of requests), the 40% of these requests came from of European countries (EU28, Norway and Switzerland), and the 60% remaining of other countries, basicly from the United States (37%). There are in Europe around 27.000 companies deploying medical tech. Around the 95% are small or medium enterprises, employing less than 50 people each.
But definitely, the critical point that marked the need of performing changes at legal issues and increase the controls was the fraud of the French company PIP (Poly Implant Prothèse), at which discovered that one of the main manufacturers of mammary implants in the world was fashioning implants with industrial silicone non surgical, after appearing numerous cases of leaks ending up in deaths by cancer. It wants , on the database facilitated by the Agency of Products of Health of France (ANSM), that there is at least some 400.000 persons affected at 65 countries, some 7.500 leaks detected and some 3.000 wives that have informed of unwanted effects, most inflammation at his implants. The company went bankrupt and sutted down in 2010.
From this scandal the European Commission created the nicknamed plan of action PIP, the origin the present Regulations focusing on:
1. Revise the operation of the organisms notified
2. Increase of the Follow-up.
3. Coordination of organisms notified and competent authorities at the case of surveillance.
4. Communication and transparency.
What is what has changed?
The impact for all the economic agents, especially for manufacturers and organisms notified is considerable since they increased the requirements:
Has capsized the classification, besides exacting, of numerous sanitary products, which thing implies main requisites at regulatory level.
They require main number of clinical data and expect more clinical enquiries, especially for the products of main risk (Class IIb and Class III).
Main requisites of follow-up.
It has created the Database EUDAMED (European Database where Medical Devices). It treats of a very ambitious project, that pretends to centralise the follow-up of all the sanitary products, since his introduction at the market until the surveillance or the clinical surveys. EUDAMED will have an open part at the public so that any unholy can consult it.
Necessity of UDI (Unique Device Identification): the traçabilitat is one of the horses of battle of the new Regulations and each sanitary product has to have a number of only identification.
Card for implants: the implants have requisites more exacting of traçabilitat, seeking the efficacy at case of alarms, or withdrawals of product. The card will be handed at the patient, hears therefore connoisseur of the manufacturer of the implant and data of traçabilitat specific.
Sufficient coverage of insurance at the companies to coat complaints of the potential harms that can cause.
It includes the figure of responsible person of fulfillment of the rule in the organisations fabricadores and of the representatives authorised.
The Regulation affects also at products with cosmetic purpose that have the same mechanism of act and therefore risks, that equivalent sanitary products, and for example lasers or squads of liposucció.
Image3: New Regulations

Source: EC.
Image 4: To keep in mind

Source: EC.
The new Regulation also involves all the implied stakeholders for a proactive attitude at the healthcare products release to the market (manufacturers, traders, sales,…). They are also garants of the compliance according to the rule.
Figura 5. New features
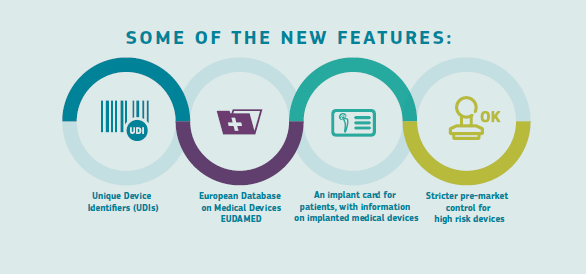
Source: EC.
The impact is considerable, implying the increase of resources invested to assure the compliance with the new regulation. To assure a softer transition, and not disregarding the market. It will enter into force gradually (image 6). Those products that need, for his typology of risk, an organism notified to obtain his labeling CE, will be able to have its products in the market while have a CE certification valid against MDD.
Image 6. Transition timeline

Font: EC.
____________________
- MDR: Medical Device Regulations
- IVDR: In Vitro Diagnostic Regulations
- MDD: Medical Device Directives
_____________________
References
- European Commission, (2020). Internal Market, Industry, Entrepreneurship and SMEs. Extracted the March 9, 2020 from https://ec.europa.eu/growth/sectors/medical-devices_en
- Agencia Española de Medicamentos y Productos Sanitarios, (2020). Productos sanitarios. Extracted the March 9, 2020 from https://www.aemps.gob.es/productos-sanitarios/productossanitarios_prodsanitarios/
- Medtech Europe, (2020). New medical technology. Extracted the March 9, 2020 from https://www.medtecheurope.org/new-medical-technology-regulations/
-
The Regulation will replace the current 93/42/EEC (MDD) (of medical devices) and the 90/385/EEC (AIMDD)